

Aisling Rayne, University of Canterbury PhD student.
For example, should we mix small, isolated populations to maximise genome-wide diversity? Or should we focus on protecting genetic diversity in locally-adapted populations? Questions such as these can inform how we move (translocate) threatened species to establish new populations or strengthen existing ones. We are increasingly looking for answers using a diverse toolbox that weaves together different knowledge systems.
Recently, the emergence of new tools in the conservation toolbox have reignited debate over whether conservation translocations should prioritise genome-wide diversity over maintaining locally-adapted variation.
To address these practical questions we first need to determine whether we can actually characterise adaptive variation, especially in threatened species. That is, can we identify and understand the genetic variation that enables species to adapt to change?
We are exploring this as part of BioHeritage project Adaptive Evolution of Native Biota – a partnership between mana whenua, conservation practitioners and researchers. We are assessing the extent to which we can characterise adaptive variation using five diverse focal species, and using the data we collect to inform culturally-responsive strategies to enhance resilience in threatened species.
Emerging genomic approaches promise to characterise adaptive variation
The first step towards characterising adaptive variation is to use DNA (or markers) to find locally-adaptive variants: that is, the genetic variants associated with traits that enable species to survive and thrive in certain environments. Thanks to high-throughput sequencing technologies—combined with rapidly dropping costs and increased capability and capacity in the conservation genetics community—we can readily produce tens to hundreds-of-thousands of markers from across the entire genome.
The next step is to combine these genomic markers with other information, such as environmental data and measures of fitness (i.e., traits associated with survival or reproduction), to identify locally-adaptive variants.
Genome-wide markers are also improving our ability to address questions previously answered using just a handful of genetic markers. For example, genomic data provide better estimates of relatedness to enhance pairing decisions in conservation breeding programmes.
There is growing interest in incorporating this additional information into conservation translocation decisions; but there are caveats. Despite a surge of interest in the characterisation of adaptive variation, there have been relatively few empirical studies to date—and fewer still have considered how to incorporate adaptive variation into conservation translocations decisions (e.g., Pacific lamprey).

Locally adapted variants are most likely to be found in well-studied, genetically diverse species
Where we see successful characterisation of adaptive variation is often in well-studied species, with a high-quality reference genome and comprehensive genomic data, as well as informative fitness measures and environmental data.
For these species, we are better able to explore a range of analytical approaches (e.g., outlier-detection based approaches, genotype-environment association studies and genome-wide association studies). Further, new studies indicate that our chances of detecting locally-adaptive variants are highest in large, connected populations that experience very different selection pressures (e.g., Nebraska deer mice).

For many threatened species it may prove challenging to characterise adaptive variation at all
Genomic approaches are more likely to include regions of the genome that are under selection compared to genetic approaches. However, it is increasingly clear that access to genomic data only forms part of the picture: whether we can detect and then characterise adaptive variation also depends on many other factors, such as population demographics, sampling approaches and availability of non-genomic data like fitness measures and environmental data.
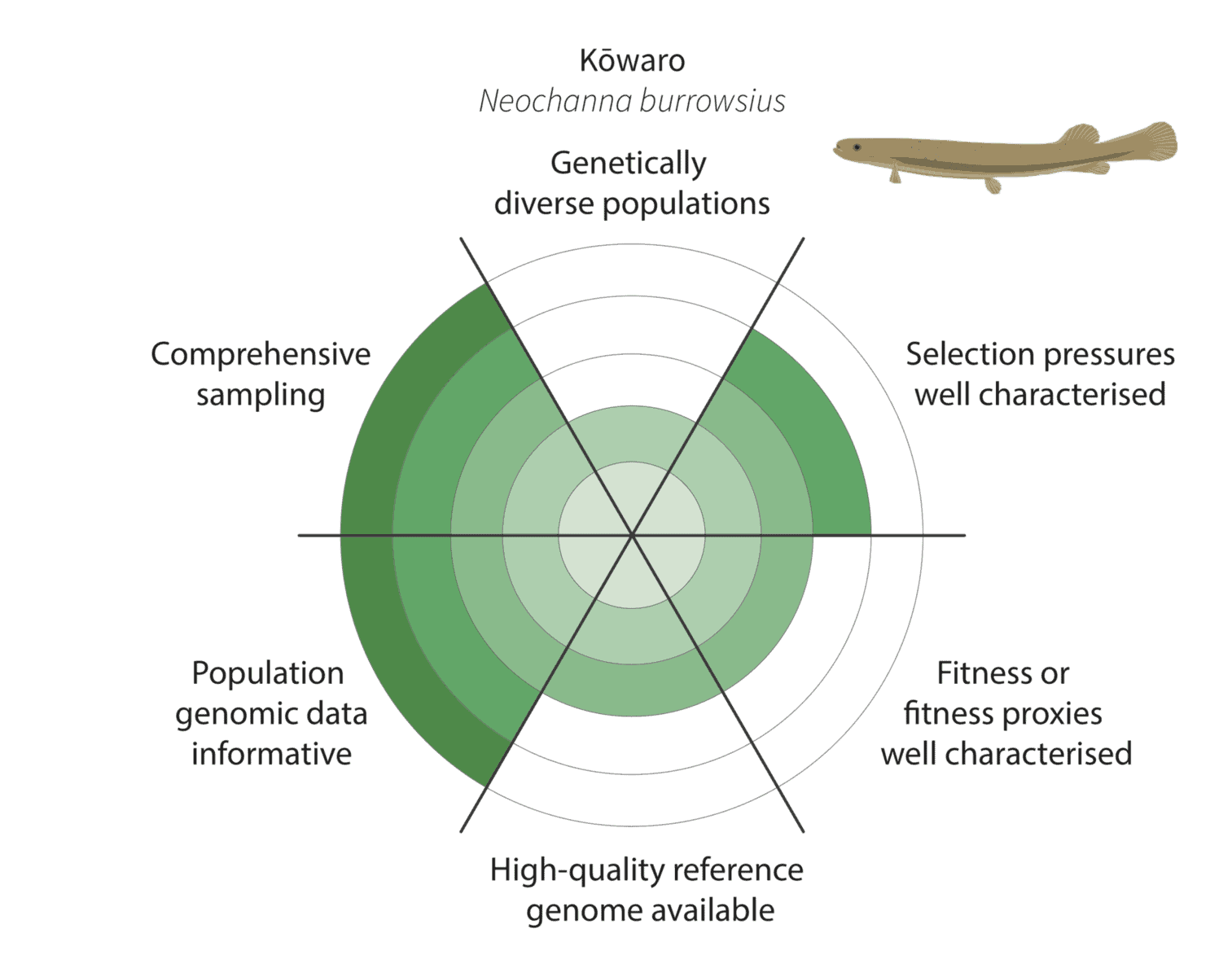
We co-developed a new framework for assessing how well these criteria have been met to enable characterisation of adaptive variation (figure below):
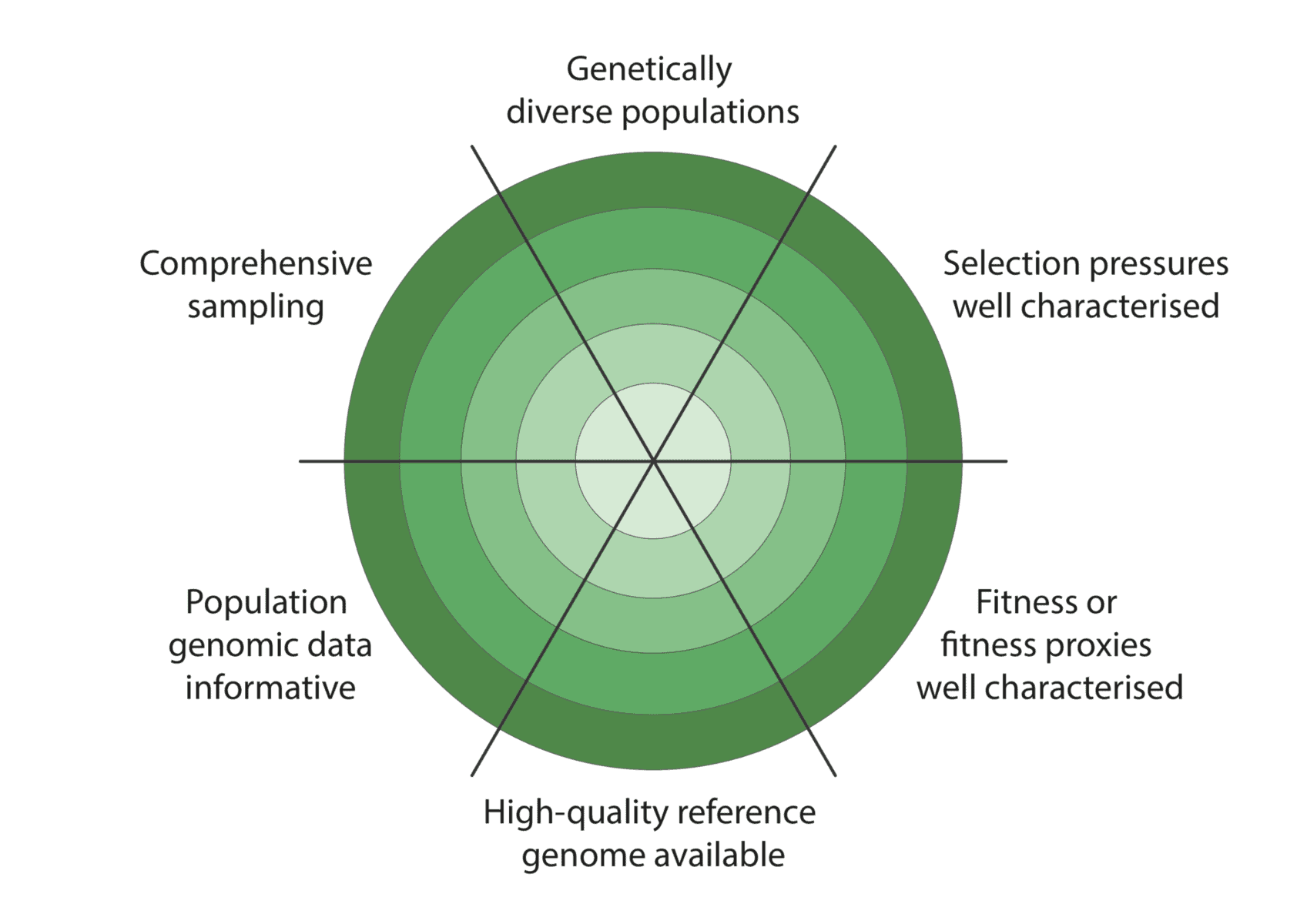
Given the constraints described above, we are being realistic about our ability to detect adaptive variation. Indeed, it may prove challenging for at least some of our focal species (figure below).
For example, for the critically endangered kōwaro (Canterbury mudfish) the coloured section extends to the circle outer edge for comprehensive sampling and population genomic data informative. This is because we are confident that most kōwaro populations can be sampled using highly informative whole genome sequencing data—and both these factors increase our chances of characterising adaptive variation.
It is more challenging for kēkēwai (freshwater crayfish) as we are less confident of the species distribution and our reduced-representation sequencing data represents only a fraction of the genome. On the other hand, we are co-generating a platinum-standard kēkēwai reference genome that will improve our ability to characterise adaptive variation.
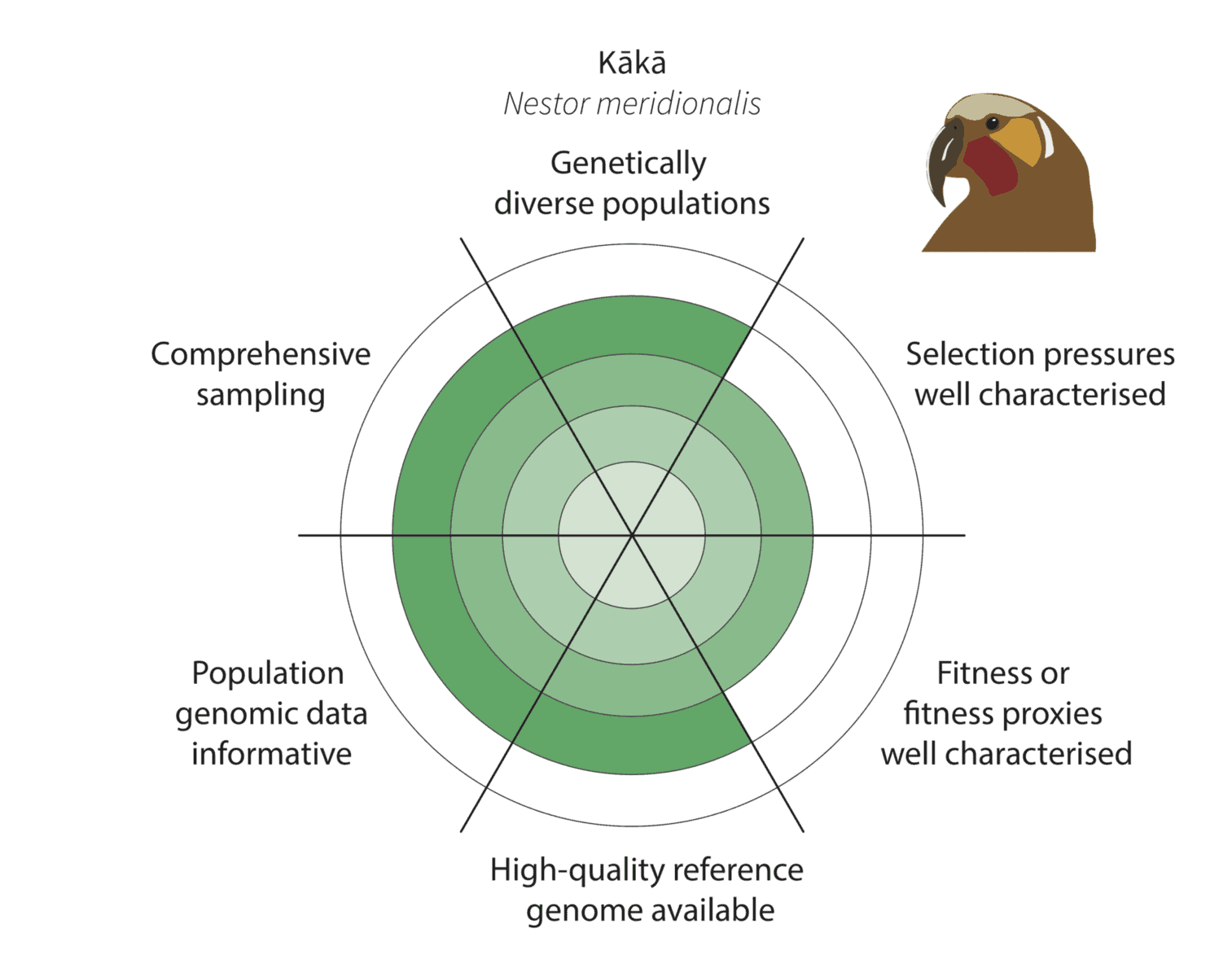
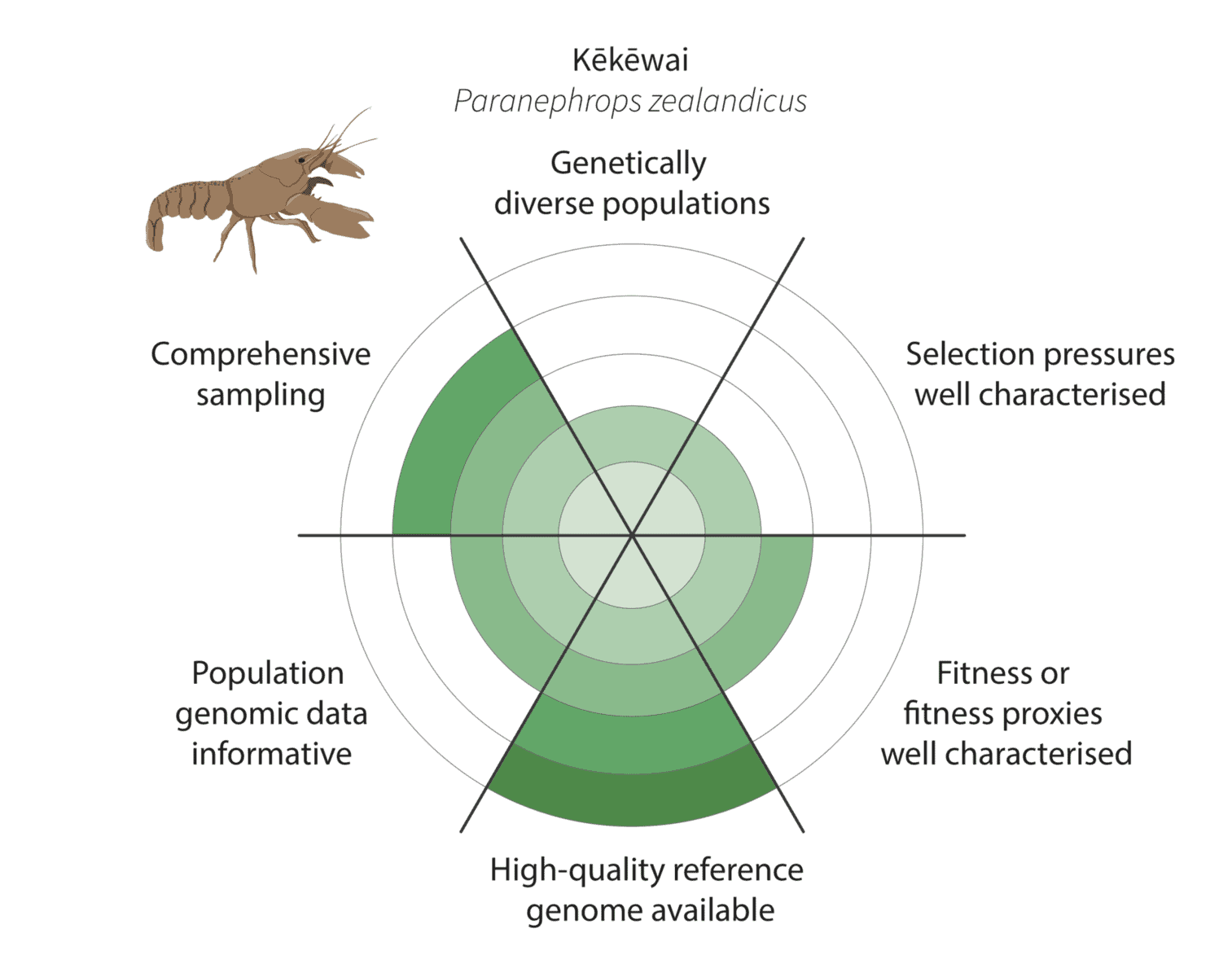
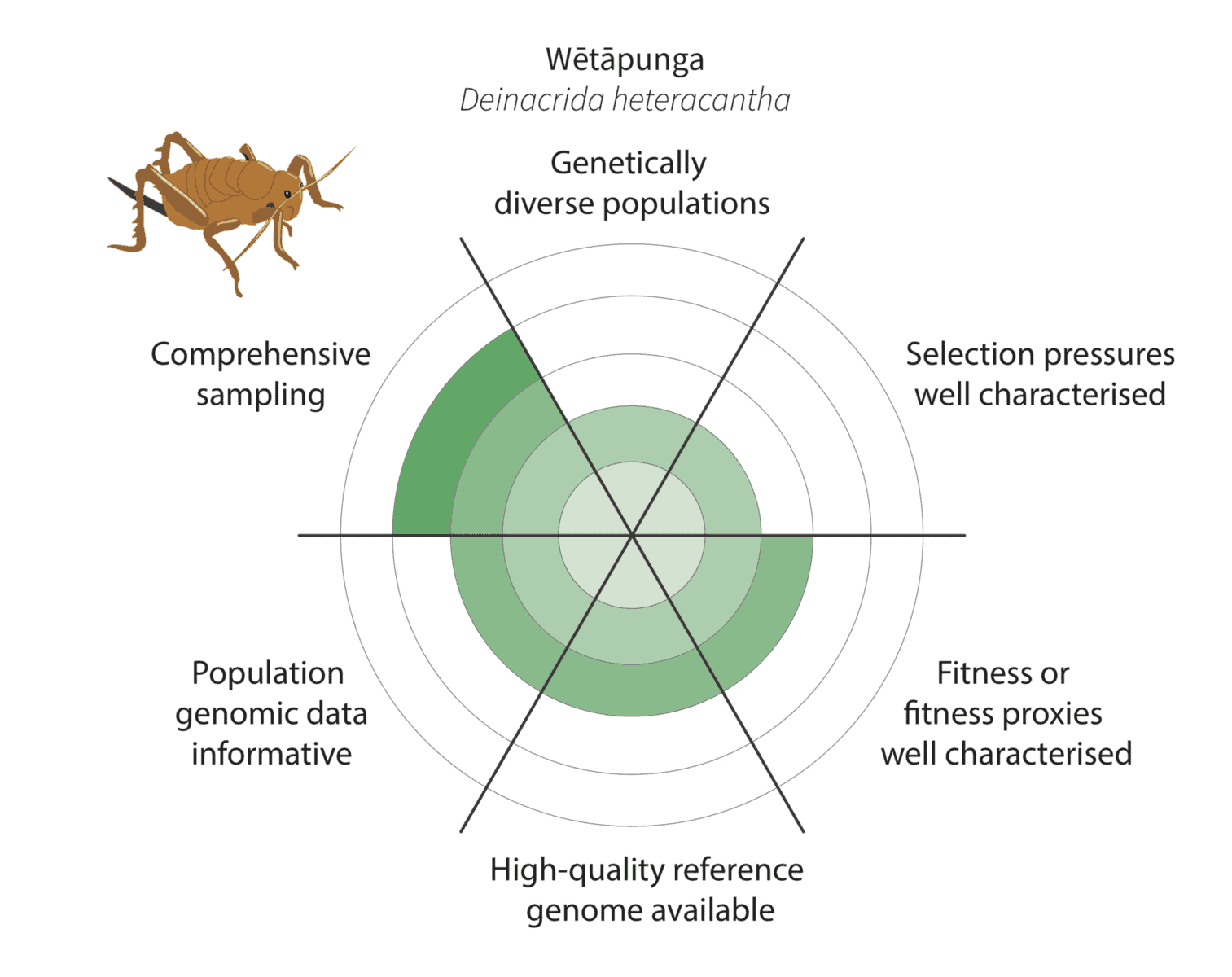
Watch this space
While characterising adaptive variation remains an exciting conservation genomics tool, it is not without its challenges. However, it is the ways in which we mitigate these challenges that will move the field forward.
We anticipate our framework will evolve as we learn more about the limitations—and opportunities—associated with characterising adaptive variation. In the meantime, to enhance resilience in Aotearoa New Zealand’s biological heritage, we will continue to co-design conservation translocations that weave genomic data into diverse ways of knowing.
The framework above was co-developed as a contribution toward an interdisciplinary review about conservation translocations in Aotearoa New Zealand in the predator-free era recently submitted to the New Zealand Journal of Ecology, and as part of the BioHeritage project Adaptive Evolution of Native Biota.